
Статья опубликована в рамках: IV Международной научно-практической конференции «Естественные и математические науки в современном мире» (Россия, г. Новосибирск, 01 апреля 2013 г.)
Наука: Химия
Секция: Физическая химия
Скачать книгу(-и): Сборник статей конференции
- Условия публикаций
- Все статьи конференции
дипломов

БЫСТРОДЕЙСТВУЮЩИЙ МЕТОД КВАНТОВОХИМИЧЕСКИХ РАСЧЕТОВ С АППРОКСИМАЦИЕЙ ФОКИАНА DFT ДЛЯ МАТЕМАТИЧЕСКОГО МОДЕЛИРОВАНИЯ ЭЛЕКТРОННОГО СТРОЕНИЯ БОЛЬШИХ МОЛЕКУЛ
Аникин Николай Алексеевич
канд. хим. наук, научный сотрудник ИОХ РАН, г. Москва
E-mail: nikan@swf.chem.ac.ru
Бугаенко Владислав Ленарович
канд. хим. наук, старший научный сотрудник ГНЦ РФ ИТЭФ, г. Москва
E-mail: bugaenko@itep.ru
Кузьминский Михаил Борисович
канд. хим. наук, старший научный сотрудник ИОХ РАН, г. Москва
E-mail:
Мендкович Андрей Семёнович
д-р хим. наук, руководитель лаборатории ИОХ РАН, г. Москва
E-mail: asm@free.net
Введение
В работе предложен новый метод квантовохимических расчетов электронной структуры больших молекул с быстродействием на уровне быстрых но менее точных полуэмпирических методов и точностью, приближенной к популярным более точным и надежным методам функционала электронной плотности (DFT, Density Functional Theory).
Предложенный квантовохимический метод NESE (“Non Empirized Semi Empirics”) [1, 2, 3] использует «моделирование» (аппроксимацию) фокиана метода DFT. В данной работе рассматривается начальная неитерационная версия метода — NESE-0. Она включает оператор кинетической энергии плюс сумму атомных потенциалов с эмпирическими параметрами, откалиброванными по МНК из отклонения от эталонного одноэлектронного фокиана DFT. Далее предполагается и количественно и качественно ее усовершенствовать, в том числе — вводя итерации метода самосогласованного поля (ССП). Работа поддержана РФФИ, проект 11-07-00470.
Численные результаты.
Метод апробирован на молекулярных системах с атомами H, C, N, O с параметрами, подобранными нами по DFT-расчетам многих тысяч разнообразных органических молекул с типичных группировками этих атомов. Метод сопоставлен как с распространенными полуэмпирическими методами AM1 [4], наиболее распространенным PM3 [6], и новейшим PM6 [7], так и с модельным методом DFT (DFTB) [8], включенным в состав известного широко употребляемого квантовохимического пакета программ Gaussian-09 [5].
Найдено (см. Рис. 1, 2, 3, 4), что радиальные функции атомных потенциалов NESE-0, как и должно быть, убывают с ростом расстояния от ядра, особенно резко — за пределами валентных контактов, приближаясь к нулю: у атома Н — после ~1,5 А, у атомов C, N, O — после ~ 2 A. Кривые для атомов водорода и углерода весьма близки для трех наборов молекул: углеводородного набора (только атомы H, C) , кислородсодержащего набора (атомы H, C, O) и азотсодержащего набора (атомы H, C, N), что свидетельствует о неплохой переносимости параметров и полезно для дальнейших применений метода.
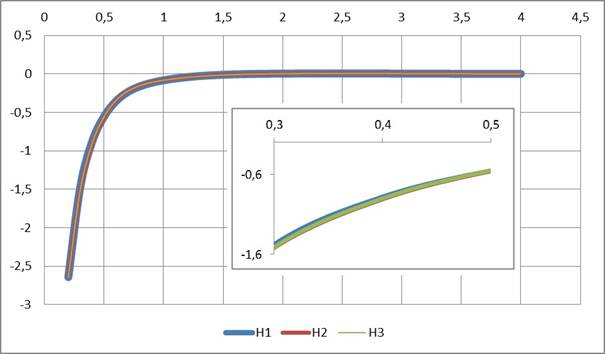
Рисунок 1. Потенциал атома водорода: H1 — оптимизированного по молекулам с атомами C,H; H2 — оптимизированного по молекулам с атомами C,H,O; H3 — оптимизированного по молекулам с атомами C,H,N

Рисунок 2. Потенциал атома углерода: C1 — оптимизированного по молекулам с атомами C,H; C2 — оптимизированного по молекулам с атомами C,H,O; C3 — оптимизированного по молекулам с атомами C,H,N
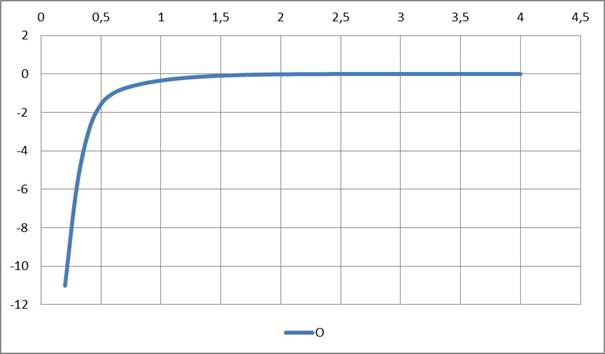
Рисунок 3. Потенциал атома кислорода
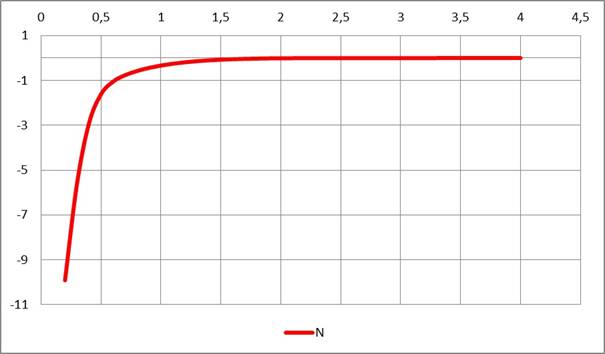
Рисунок 4. Потенциал атома азота
Найдено, что уже нынешняя неитерационная версия NESE-0 предлагаемого метода кардинально (по порядку величины) точнее итерационных полуэмпирических методов (AM1, PM3, PM6) воспроизводит орбитальные энергии валентных уровней эталонного метода DFT (функционал PBE в базисе MINI, PBE/MINI), и превосходит точность (хотя и не столь кардинально) итерационного метода DFTB, особенно по зазору между верхними занятыми и нижними вакантными МО, что особенно важно при рассмотрении вариации (возмущении) электронной структуры молекул. Превосходство предлагаемого метода NESE-0 носит систематический характер.
Углеводородные молекулы.
Например, на 2960 молекулах углеводородов (включая сложные пи-сопряженные и полициклические) отклонение от эталонного DFT составило:
·у предлагаемого неитерационного метода NESE-0: RMS 0,026308 a. e., средняя величина отклонения (в одну сторону) — 0,010155 a. e., максимальное отклонение 0,131486 a. e.;
·у итерационного полуэмпирического метода AM1: RMS 0,27485, среднее одностороннее — 0,22987 , максимальное отклонение 0,92797 a. e.;
·у итерационного полуэмпирического метода PM3: RMS 0,27212 a. e., среднее одностороннее — 0,23066, максимальное отклонение 0,90352 a. e.;
·у итерационного полуэмпирического метода PM6: RMS 0,21552 a. e. среднее одностороннее — 0,18706, максимальное отклонение 0,91536 a. e.;
·у итерационного метода DFTBa, моделирующего DFT: RMS 0,12349 a. e, среднее одностороннее 0,05672, максимальное отклонение 1,32266 a. e.
Точность (RMS) описания HOMO (верхних занятых МО): NESE-0: 0,026105, AM1: 0,13465, PM3: 0,13982, PM6: 0,13263, DFTBa: 0,01563 a. e.
Точность (RMS) LUMO (нижних вакантных МО): NESE-0: 0,026525, AM1: 0,07643, PM3: 0,07934, PM6: 0,07016, DFTBa: 0,03223 a. e.
Точность (RMS) зазора между LUMO и HOMO: NESE-0: 0,011641, AM1: 0,14995, PM3: 0,14843, PM6: 0,15091, DFTBa: 0,02046 a. e.
Видно, что точность предлагаемого неитерационного метода NESE-0 примерно в 10 раз превышает точность полуэмпирических методов (AM1, PM3, PM6), которые в среднем мало отличаются друг от друга (хотя на отдельных молекулах они отличаются значительно). По сравнению с итерационным метода DFTBa, моделирующего DFT, NESE-0 выигрывает меньше, в 2—4 раза.
Точность довольно равномерна по разным группам данного класса молекул. Так, для 715 разнообразных молекул алкенов и полиенов, включая циклические, отклонение от эталонного DFT составило:
·у предлагаемого метода NESE-0: RMS 0,022544 a. e. (у алканов и циклоалканов средняя величина RMS была 0,014931 a. e.), отклонения (в одну сторону) — 0,010189, максимальное отклонение 0,095188 a. e.;
·у метода AM1: RMS 0,26871, среднее в одну сторону — 0,23201, максимальное отклонение 0,68863 a. e.;
·у метода PM3: RMS 0,27049 a. e., среднее в одну сторону — 0,18813, максимальное отклонение 0,78107 a. e.;
·у метода PM6: RMS 0,20756 a. e. среднее одностороннее — 0,18706, максимальное отклонение 0,67538 a. e.;
·у метода DFTBa, моделирующего DFT: RMS 0,08477 a. e., среднее одностороннее 0,03585, максимальное отклонение 0,70856 a. e.
Точность зазора LUMO-HOMO: NESE-0: 0,006406, AM1: 0,17550, PM3: 0,17278, PM6: 0,17743, DFTBa: 0,02059 a. e.
Здесь также выигрыш NESE-0 у полуэмпирических методов на порядок, а у DFTB — в 2—4 раза.
Кислородсодержащие молекулы.
Точность на 2846 сложных и разнообразных молекулах с атомами углерода, водорода и кислорода (включая гликозиды и т. д.) составила:
·у предлагаемого метода NESE-0: RMS 0,022926 a. e., средняя величина отклонения — 0,007262, максимальное отклонение 0,133307 a. e.;
·у метода AM1: RMS 0,27045, среднее одностороннее — 0,23113, максимальное отклонение 0,92101 a. e.;
·у метода PM3: RMS 0,26505, среднее одностороннее — 0,22945, максимальное отклонение 0,90282 a. e.;
·у метода: RMS 0,21397, среднее одностороннее — 0,19098, максимальное отклонение 0,90858 a. e.;
·у метода DFTBa: RMS 0,12029 a. e., среднее одностороннее 0,06000, максимальное отклонение 1,32266 a. e.
Точность зазора LUMO-HOMO: NESE-0: 0,017913, AM1: 0,18242, PM3: 0,18155, PM6: 0,17022, DFTBa: 0,06288 a. e.
Точность близка в точности для углеводородов, в среднем немного больше погрешность зазора LUMO-HOMO, что естественно ввиду значительной полярности связей C-O и O-H.
Азотсодержащие молекулы.
Точность на 2035 сложных и разнообразных молекулах с атомами углерода, водорода и азота (включая пи-сопряженные гетероциклы) составила:
·у предлагаемого метода NESE-0: RMS 0,026409 a. e., среднее одностороннее отклонение — 0,009705, максимальное отклонение 0,129880 a. e.;
·у метода AM1: RMS 0,27178, среднее одностороннее — 0,22832, максимальное отклонение 0,92101 a. e.;
·у метода PM3: RMS 0,26505, среднее одностороннее — 0,22591, максимальное отклонение 0,90282 a. e.;
·у метода PM6: RMS 0,21574, среднее одностороннее — 0,18849, максимальное отклонение 0,90858 a. e.;
·у метода DFTBa: RMS 0,12853 a. e., среднее одностороннее 0,06298, максимальное отклонение 1,27607 a. e.
Точность (RMS) зазора LUMO-HOMO: NESE-0: 0,019456, AM1: 0,17202, PM3: 0,16629, PM6: 0,16892, DFTBa: 0,02876 a. e.
Точность близка к точности предыдущих групп.
Обсуждение.
В дальнейшем предложенный метод NESE-0 предполагается усовершенствовать за счет неитерационного учета влияния окружения данного атома на его потенциал и учета анизотропии потенциала атома (это было особенно видно на полиацетиленах, где сильна анизотропия вдоль цепи атомов углерода). Этот неитерационный метод (в усовершенствованной версии) может быть полезен:
·для расчетов мало полярных органических молекул или молекул с изолированными полярными связями без их включения в цепь пи-сопряжения (успешность этого продемонстрирована на кислород- и азот-содержащих молекулах);
·как хорошее начальное приближение для итерационных методов с любым заданным функционалом DFT;
·в методах класса QM/QM с заданным функционалом DFT, где будет отсутствовать резкое отличие методов (например, NESE-0/PBE), так как NESE-0 как раз и моделирует данный функционал DFT.
Также предполагается сооружение итерационной версии NESE, но при сохранении экономичности метода на уровне DFTB и близко к полуэмпирическим методам.
Выводы.
Предложенный принципиально новый метод NESE (“Non Empirized Semi Empirics”), моделирующий расчеты точными, но ресурсоемкими методами DFT; определены пути его дальнейшего развития.
Результаты апробации начальной версии метода NESE-0 на многих тысячах разнообразных органических молекул содержащих атомы C, H, N, O, показывают, что даже эта начальная несовершенная и неитерационная версия метода по точности моделирования фокиана (Гамильтониана) DFT умеренно превосходит итерационный метод DFTB, моделирующий DFT, и на порядок — итерационные полуэмпирические методы AM1, PM3, PM6 (известные методы сходного уровня ресурсоемкости).
Использование метода NESE, в том числе и в комбинации с базовыми методами DFT, может быть актуально для проведения массовых расчетов больших молекул (наноструктур и биомолекул), в т. ч. для задач конструирования лекарственных средств, где применение DFT приводит к недопустимо высоким затратам вычислительных ресурсов, а полуэмпирические методы не могут быть использованы из-за недостаточной точности и надежности.
Список литературы:
1.Аникин Н.А., Кузьминский М.Б. Новая аппроксимация DFT для быстрых расчетов био- и наноструктур // Информационно-вычислительные технологии в науке — 2011, Сб. тезисов. — М.: 2011 — С. 29—30.
2.Anikin N.A., Bugaenko V.L., Anisimov V.M. Chemically parameterized fast DFT method // 225th ACS National Meeting (New Orleans)/COMP / Computers in Chemistry. P. 243.
3.Anikin N.A., Bugaenko V.L., Anisimov V.M. Semi-DFT approach to calculation of protein systems // 226th ACS National Meeting (New York, Fall 2003)/COMP / Computers in Chemistry. P. 112.
4.Dewar M.J.S., Zoebisch E.G., Healy E.F., Stewart J.J.P. Development and use of quantum mechanical molecular models. 76. AM1: A new general purpose quantum mechanical molecular model // J. Am. Chem. Soc. 1985. Vol. 107. № 13. P. 3902—3909.
5.Frisch M.J., Trucks G.W., Schlegel H.B., e. a. Gaussian 09, Revision A.02 // Gaussian, Inc., Wallingford CT. 2009.
6.Stewart J.J.P. Optimization of parameters for semiempirical methods I. Method // J. Comput. Chemistry. 1989. Vol. 10. № 2. P. 209—220.
7.Stewart J.J.P. Optimization of Parameters for Semiempirical Methods V: Modification of NDDO Approximations and Application to 70 Elements // J. Molec. Modeling. 2007. Vol. 13. № 12. P. 1173—1213.
8.Zheng G., Lundberg M., Jakowski J., Vreven T., Frisch M.J., Morokuma K. Implementation and benchmark tests of the DFTB method and its application in the ONIOM method // Int. J. Quant. Chem. 2009. Vol. 109. № 9. P. 1841—1854.
дипломов